Annotating cell clusters by integrating single-cell RNA-seq data#
Introduction#
In this tutorial we will integrate single-cell RNA-seq data to annotate the cells in our ATAC-seq data. Before we begin, you should have read the Standard Pipeline tutorial.
In addition to SnapATAC2, we will utilize scanpy and scvi-tools to perform the integration.
[1]:
import warnings
warnings.filterwarnings("ignore")
import snapatac2 as snap
import anndata as ad
import pandas as pd
import scanpy as sc
import scvi
import numpy as np
scvi.settings.seed = 0
snap.__version__
2023-09-10 13:46:23 - INFO - Created a temporary directory at /scratch/kaizhang/33240354.tscc-mgr7.local/tmpy92jeahy
2023-09-10 13:46:23 - INFO - Writing /scratch/kaizhang/33240354.tscc-mgr7.local/tmpy92jeahy/_remote_module_non_scriptable.py
INFO: Global seed set to 0
2023-09-10 13:46:27 - INFO - Global seed set to 0
[1]:
'2.3.2dev1'
Preparing data#
We first import the reference single-cell RNA-seq data in which the cells have been annotated.
Note here we use backed=None to load the AnnData object into memory, so that it can be used by scanpy and scvi-tools.
[2]:
reference = snap.read(snap.datasets.pbmc10k_multiome(), backed=None)
reference
[2]:
AnnData object with n_obs × n_vars = 9631 × 29095
obs: 'domain', 'cell_type'
var: 'gene_ids', 'feature_types'
We then import the cell-by-bin matrix of single-cell ATAC-seq data and use the snap.pp.make_gene_matrix to generate gene activity matrix.
[3]:
atac = snap.read(snap.datasets.pbmc5k(type='h5ad'), backed=None)
query = snap.pp.make_gene_matrix(atac, gene_anno=snap.genome.hg38)
query
[3]:
AnnData object with n_obs × n_vars = 4436 × 60606
obs: 'tsse', 'n_fragment', 'frac_dup', 'frac_mito', 'doublet_probability', 'doublet_score', 'leiden'
Finally, we merge reference data and query data together and use the scanpy library to find out highly variable genes. After this, we are ready to utilize scvi-tools to perform the integration.
[4]:
query.obs['cell_type'] = pd.NA
data = ad.concat(
[reference, query],
join='inner',
label='batch',
keys=["reference", "query"],
index_unique='_',
)
data
[4]:
AnnData object with n_obs × n_vars = 14067 × 20169
obs: 'cell_type', 'batch'
[5]:
sc.pp.filter_genes(data, min_cells=5)
sc.pp.highly_variable_genes(
data,
n_top_genes = 5000,
flavor="seurat_v3",
batch_key="batch",
subset=True
)
Data integration#
First we setup the scvi-tools to pretrain the model.
[6]:
scvi.model.SCVI.setup_anndata(data, batch_key="batch")
vae = scvi.model.SCVI(
data,
n_layers=2,
n_latent=30,
gene_likelihood="nb",
dispersion="gene-batch",
)
2023-09-10 13:47:04 - INFO - Unable to initialize backend 'cuda': FAILED_PRECONDITION: No visible GPU devices.
2023-09-10 13:47:04 - INFO - Unable to initialize backend 'rocm': NOT_FOUND: Could not find registered platform with name: "rocm". Available platform names are: Interpreter Host CUDA
2023-09-10 13:47:04 - INFO - Unable to initialize backend 'tpu': module 'jaxlib.xla_extension' has no attribute 'get_tpu_client'
2023-09-10 13:47:04 - INFO - Unable to initialize backend 'plugin': xla_extension has no attributes named get_plugin_device_client. Compile TensorFlow with //tensorflow/compiler/xla/python:enable_plugin_device set to true (defaults to false) to enable this.
2023-09-10 13:47:04 - WARNING - No GPU/TPU found, falling back to CPU. (Set TF_CPP_MIN_LOG_LEVEL=0 and rerun for more info.)
[7]:
vae.train(max_epochs=1000, early_stopping=True)
INFO: GPU available: False, used: False
2023-09-10 13:47:05 - INFO - GPU available: False, used: False
INFO: TPU available: False, using: 0 TPU cores
2023-09-10 13:47:05 - INFO - TPU available: False, using: 0 TPU cores
INFO: IPU available: False, using: 0 IPUs
2023-09-10 13:47:05 - INFO - IPU available: False, using: 0 IPUs
INFO: HPU available: False, using: 0 HPUs
2023-09-10 13:47:05 - INFO - HPU available: False, using: 0 HPUs
Epoch 462/1000: 46%|██████████████████████████████ | 462/1000 [14:09<16:28, 1.84s/it, v_num=1, train_loss_step=2.36e+3, train_loss_epoch=2.56e+3]
Monitored metric elbo_validation did not improve in the last 45 records. Best score: 2627.320. Signaling Trainer to stop.
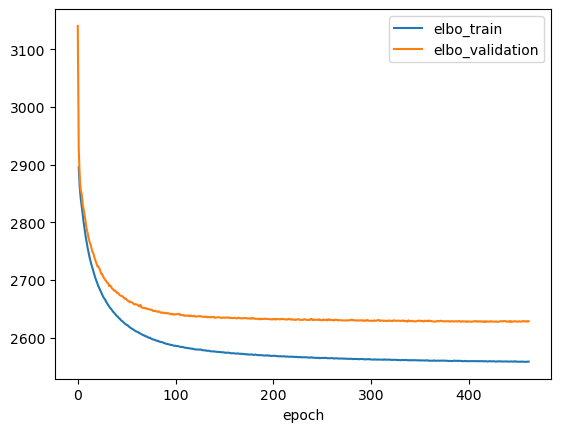
Let’s plot the training history and make sure the model has converged.
[8]:
ax = vae.history['elbo_train'][1:].plot()
vae.history['elbo_validation'].plot(ax=ax)
[8]:
<AxesSubplot: xlabel='epoch'>
[9]:
data.obs["celltype_scanvi"] = 'Unknown'
ref_idx = data.obs['batch'] == "reference"
data.obs["celltype_scanvi"][ref_idx] = data.obs['cell_type'][ref_idx]
[10]:
lvae = scvi.model.SCANVI.from_scvi_model(
vae,
adata=data,
labels_key="celltype_scanvi",
unlabeled_category="Unknown",
)
[11]:
lvae.train(max_epochs=1000, n_samples_per_label=100)
INFO Training for 1000 epochs.
INFO: GPU available: False, used: False
2023-09-10 14:01:15 - INFO - GPU available: False, used: False
INFO: TPU available: False, using: 0 TPU cores
2023-09-10 14:01:15 - INFO - TPU available: False, using: 0 TPU cores
INFO: IPU available: False, using: 0 IPUs
2023-09-10 14:01:15 - INFO - IPU available: False, using: 0 IPUs
INFO: HPU available: False, using: 0 HPUs
2023-09-10 14:01:15 - INFO - HPU available: False, using: 0 HPUs
Epoch 1000/1000: 100%|███████████████████████████████████████████████████████████████| 1000/1000 [51:09<00:00, 3.07s/it, v_num=1, train_loss_step=2.67e+3, train_loss_epoch=2.63e+3]
INFO: `Trainer.fit` stopped: `max_epochs=1000` reached.
2023-09-10 14:52:25 - INFO - `Trainer.fit` stopped: `max_epochs=1000` reached.
Epoch 1000/1000: 100%|███████████████████████████████████████████████████████████████| 1000/1000 [51:09<00:00, 3.07s/it, v_num=1, train_loss_step=2.67e+3, train_loss_epoch=2.63e+3]
[12]:
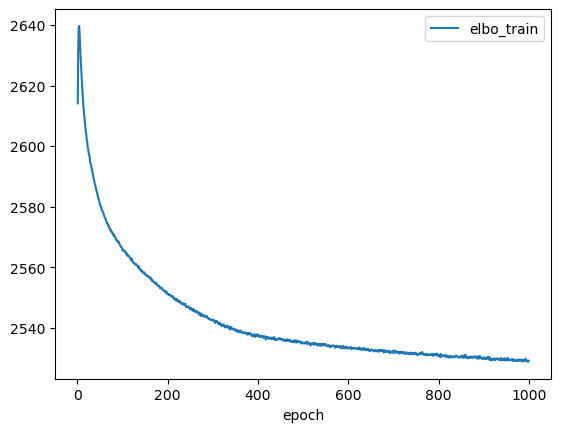
lvae.history['elbo_train'][1:].plot()
[12]:
<AxesSubplot: xlabel='epoch'>
We now can perform the label transfer/prediction and obtain the joint embedding of reference and query data.
[13]:
data.obs["C_scANVI"] = lvae.predict(data)
data.obsm["X_scANVI"] = lvae.get_latent_representation(data)
[14]:
sc.pp.neighbors(data, use_rep="X_scANVI")
sc.tl.umap(data)
[15]:
sc.pl.umap(data, color=['C_scANVI', "batch"], wspace=0.45)
... storing 'celltype_scanvi' as categorical
... storing 'C_scANVI' as categorical

Save the predicted cell type labels back to the original cell by bin matrix.
[16]:
atac.obs['cell_type'] = data.obs.loc[atac.obs_names + '_query']['C_scANVI'].to_numpy()
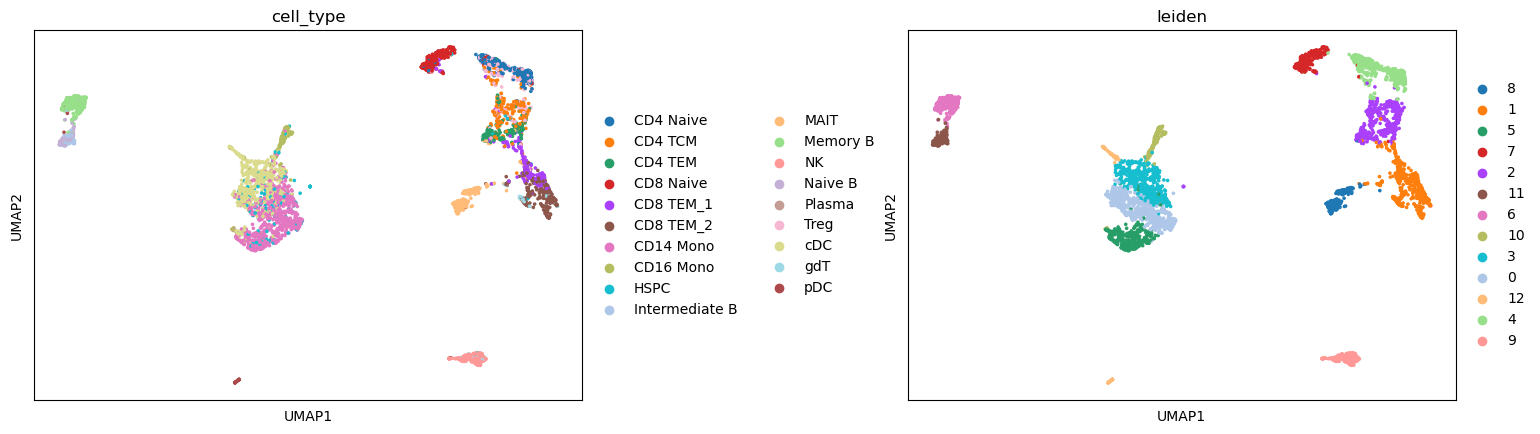
We can see that the predicted cell type labels are pretty consistent with the leiden cluster labels. Since our ATAC-seq data has fewer cells, we do not have the power to separate CD8 T and CD4 T cells, as well as a few other subtypes.
[17]:
sc.pl.umap(atac, color=['cell_type', "leiden"], wspace=0.45)
... storing 'cell_type' as categorical
Now let’s save the annotated AnnData object for future use.
[18]:
atac.write("pbmc5k_annotated.h5ad", compression="gzip")